Golden rule : Water tends to follows sodium
This is one topic where my boss tells me everyone remembers it and believe they have finally understand it for that few seconds – then POOF, it is gone out of the mind almost immediately.
I so TOTTALLY agree.
I mean, who can be bothered to remember all these details about random stuff?!??! There is so much more important things to do! But remember I must and recall it I will.
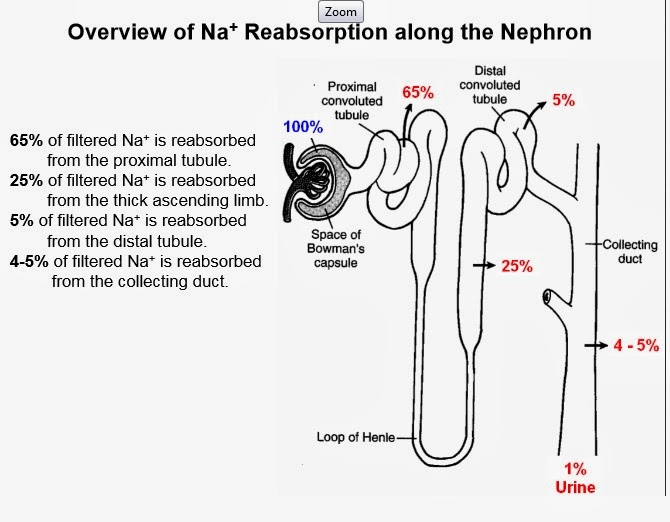
I’m someone who gets confused easily. Different terms and english names means not much to me to be honest. Apical? Basolateral? Up, down, left, right? Whatevs. I’ll simplify it for the sake of my space constrained brain.
So what we are really interested in in tubular pathology is where sodium is reabsorbed, what we can do to modify it And all the genetic deficiencies of certain transporters that leads to all these RARE diseases.
I’ll do my best to make it simple. Let’s start with the basic renal epithelial cell.
So random terms like apical membrane denotes the surface of the cell that is in contact with the urine, and basolateral membrane is the surface with blood.
Comprehende?
Few things to note:
- The apical membrane can vary its permeability to allow Na+ in
- It can have other transporters on its surface to allow movement of solutes in and out of the cell up and down the concentration gradient.
- Na+ movement is really INEFFICIENT
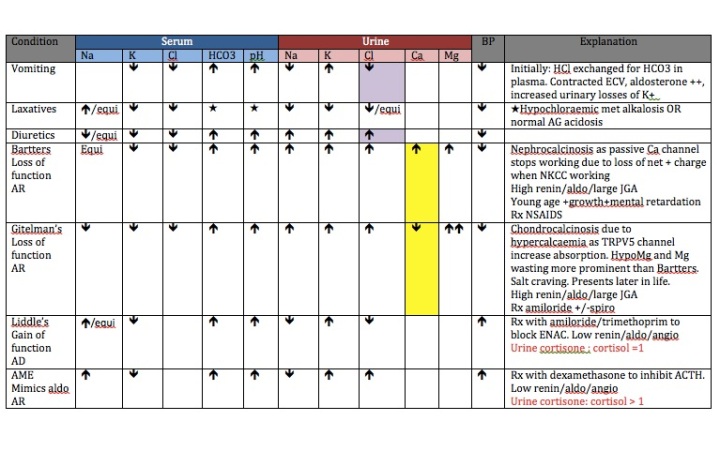
So moving down the glomerulus, we first encounter the proximal tubule.
The Proximal Tubule consists of 2 parts:
- Convulated tubule
- Descending loop of Henle
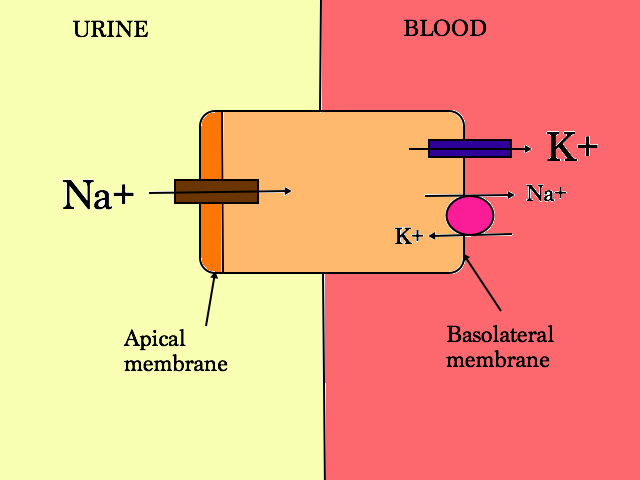
The PCT has 3 main functions:
- Bicarbonate excretion
- via carbonic anhydrase (carbonic anhydrase inhibitors eg acetazolamide works here)
- Na+ dependant mechanism
- Proteins reabsorption
- Reabsorbed via apical membrane
- endocytosed in cell
- Glucose, amino acids and phosphate is reabsorbed here and if not, it is lost forever
- Drug transport
- a lot of drugs are excreted in the proximal tubules
- Manipulation of the PCT can be achieved for therapeutic purposes
- e.g. to increase penicillin concentration in the serum, probenecid blocks the organic anion transporter inhibiting excretion of penicillin
- Trimethoprim blocks the organic anion transporter too, competing with creatinine secretion.
Problems with the PCT
Fanconi Syndrome
- global breakdown of proximal tubule transport
- loss into urine of
- phosphate -> rickets/osteomalacia & bone pain
- aa
- LMW proteins
- glucose -> glycosuric but normal plasma glucose concentration
- bicarbonate -> metabolic acidosis (proximal RTA)
- K+
- Vitamin D is not hydroxylated
- Pathogenesis?
- apical membrane abnormality
- energetic failure of ATP
- back leak of solutes

In children:
- growth failure
- episodes of hypovolaemia (polyuria due to loss of concentrating ability)
- rickets/osteomalacia
- constipation
- muscle weakness (hypokalaemia)
Special Mention:
Dent’s Disease
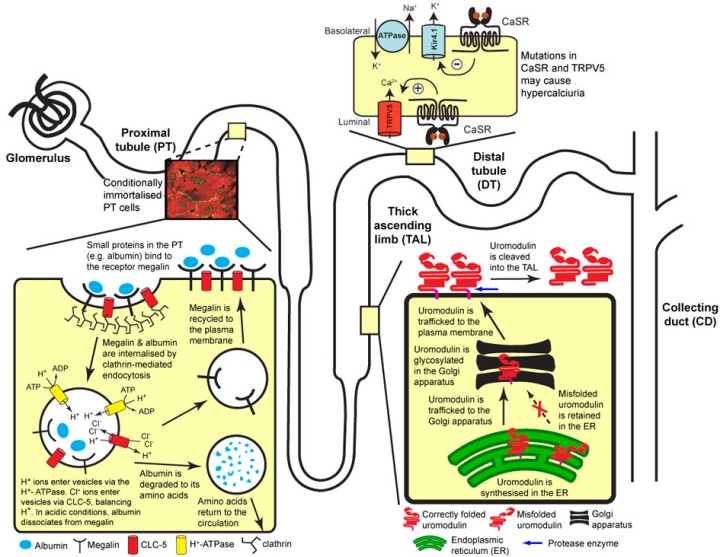
- X-linked recessive: 2 types: Type 1 ( mutation in CLCN5; Chloride voltage channel 5 gene), Type 2 (missense mutation in ORCL gene; also encodes Lowe Syndrome. Lowe syndrome sufferers have cataracts, RTA and mental retardation (oculocererbrorenal syndrome) which Dent sufferers do not have)
- CLCN5 codes for ClC5 (a H+/Cl- exchanger) in the endosome of the PT – its role is to maintain pH reabsorption of amino acids and proteins.
- ORCL gene codes for an enzyme that regulates phosphatidylinisitol 4,5 – diphosphate that is part of the membrane phospholipid. The ORCL enzyme is also found in the kidney endosome and in the actin skeleton and primary cilia
Sufferers get:
- Fanconi syndrome
- Tubular proteinuria
- Hypercalciuria, leading to
- Nephrocalcinosis and kidney stones
- CKD
Proximal Tubule Acidosis (Type 2 RTA)
- Failure to reabsorb bicarbonate
- 3 types that can all present with Fanconi’s
- Autosomal recessive with ocular abnormalities
- Autosomal recessive with osteopetrosis and cerebral calcification (inherited carbonic anhydrase II deficiency)
- Autosomal dominant
In adults:
What is it:
- HCO3 reabsorptive failure
- Lowering of threshold for HCO3 absorption in proximal tubule
- Distal acidification intact
- Can lower pH <5.5 (when given ammonium chloride test)
- Isolated or Fanconi
Causes
Primary (isolated)
- AD
- AR – SLC4A4 (Na-HCO3 transport)
- Short stature
- Ocular abnormalities
Secondary
- Familial
- Cystinosis
- AR – mutation in CTNS gene
- Lysosomal transporter disease
- Cystine accumulates in cells of PT –> toxic
- Presents in infancy
- Extra renal manifestations:
- Corneal deposits
- Hypothyroidism
- Growth retardation
- Hypophosphataemic rickets
- Tyrosinaemia
- Wilson’s disease
- Lowe Syndrome (XLR, oculocerebralrenal syndrome)
- Type 1 glycogen storage disease (Von Gierke)
- Cystinosis
- Monoclonal gammopathies
- Drugs
- Tenofovir
- Ifosfamide
- CA inhibitors (topiramate)
- Heavy metals
- Lead
- Cadmium
- Mercury
- Sjogrens
- PNH
Will lose LMW proteins
- Albustix neg
- uACR normal
- uPCR ↑
- Usu <2g/d
Can measure:
- RBP:cr
- NAG:Cr
Clinical phenotype in acquired adult disease predominantly bone (osteomalacia)
- HCO3 12-20 mmol/l
- Urine pH variable
- >5.5 if given alkali (bicarbonaturia)
- <5.5 when [HCO3] at threshold (ammonium acidification tests causes consumption of HCO3 when ammonia is converted to urea in the liver; in T2RTA, urine can be acidified further at the distal tubule hence urine pH can fall < 5.5)
- Urine AG NEGATIVE (nb direct NH4+ measurement)
OK.
Moving down the nephron, we next encounter the thick ascending limb (TAL).
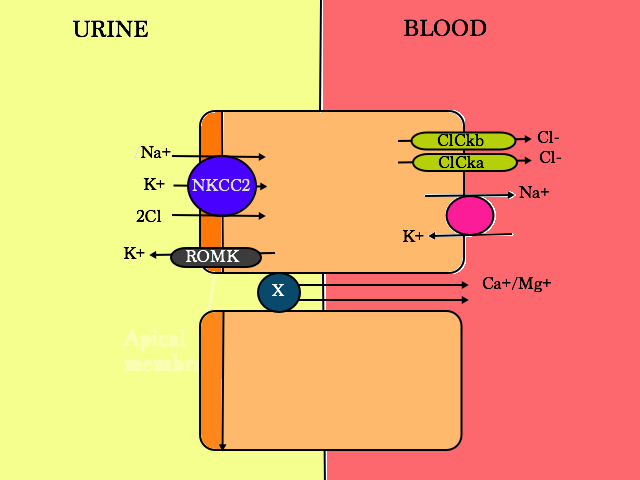
Probably by favourite cell in the nephron.
Pretty self explanatory, 25% of Na is reabsorbed here so this is where we try to stop that from happening. It is also where the weird and wonderful problem known as Bartter Syndrome starts.
Loop diuretics – knock out NKCC2, decreased Na+ reabsorption into the blood, hence H2O moves into the urine via osmosis (with Na+)
Bartters Syndrome
Recessive mustations of:
- SLC 12 A1 gene, codes for NKCC2 : Bartter Type 1 – a primary defect of SALT wasting.
- Loss of water from the body +++ –> hypotensive (low BP)
- Hypotension activates aldosterone
- Aldosterone activates the transcription of more ENAC channels at the apical membrane in the intercalated cells of the Collecting Duct to retain more Na+ in the blood, but of course, K+ is lost –> hypo K+
- Loss of K+ into the urine –> intracellularly, H+ accumulation occurs to maintain electro-neutrality. Therefore, less H+ in the serum –> metabolic alkalosis (when you lose K+, you lose H+ too)
- Ca2+/Mg2+ is reabsorbed as there is a net positive charge in the urine when NKCC is working properly (as K+ diffuses freely across to the urine when 2 Cl- are reabsorbed into the blood via the Cl- channels ). When ROMK is not working, positive charge in the lumen reduces and X transporter stops working, hence more Ca2+ is lost in the urine –> hypercalciuria & nephrocalcinosis
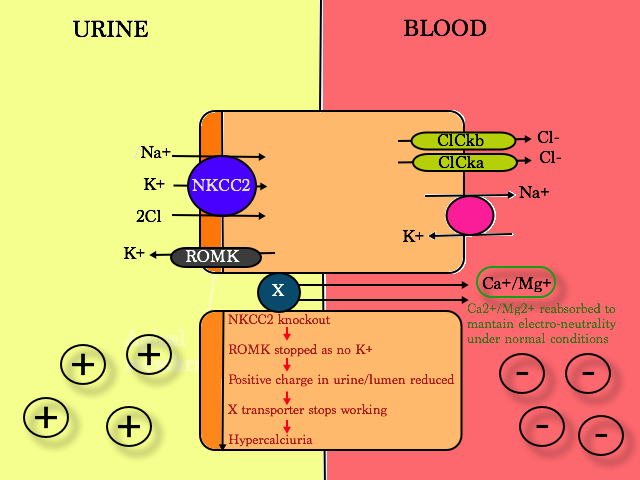
2. KCNJ1 gene, coding for the K+- voltage gated channel; i.e. ROMK knockout ; Bartter Type 2
3. Cl Ckb gene, coding for the chloride-voltage gated channel Kb; i.e. Cl Ckb knockout; Bartter Type 3. Also present in DCT – overlap with Gitelman’s
5. BSND and Cl Ckb/ClCka gene – BSND : Bartin Cl Ck beta accessory subunit ; Bartter Type 4a, and the ClCka/b channel knockout – Bartter Type 4b
Types 1, 2 and 4 are present from birth. Type 4 is also associated with sensorineural deafness as ClCk gene is also present in the ear.
Children present with:
- Failure to grow
- Nocturnal eneuresis
- Weakness
- Polyuria
- Low IQ
Histology : JGA hyperplasia
Rx : NSAIDs
Special Mention
FHHNC – Familial hypomagnesemia, hypercalciuria and nephrocalcinosis
- AR
- Mutations in CLDN 16 and CLDN19
- Hypercalciuric & HypoMg –> Nephrocalcinosis –> Renal failure
Moving further down the nephron, we now arrive at the Distal convulated tubule which gets tubular glomerular feedback from the juxtaglomerular apparatus;
The DCT
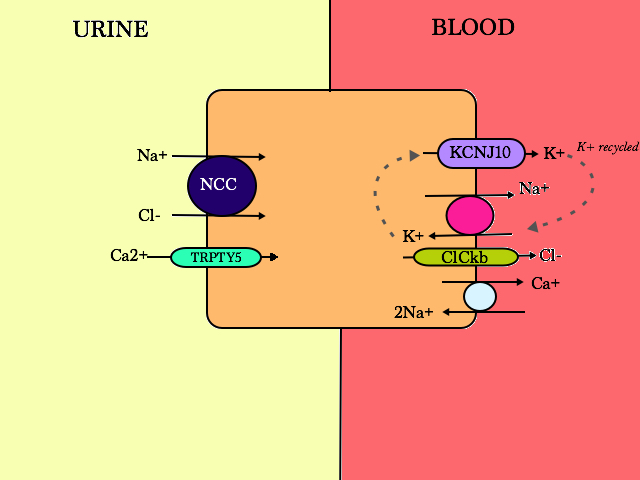
The DCT senses Na/Cl in the filtrate via the NCC receptor
On the apical membrane, NCC absorps 1 Na+ with 1 Cl-, making the process electroneutral. Loss of function of the NCC transporter causes Gitelman Syndrome (familial hypokalaemia-hypomagnesaemia):
- SLC 12A3 gene, recessive mutation
- Loss of Na –> water follows Na –> hypovolaemia –> normal or low BP
- Low BP activates aldosterone (and renin and angiotensin)
- Aldosterone activates the transcription of more ENAC channels at the apical membrane of the intercalated cells of the Collecting Duct to retain more Na+ in the blood, but of course, K+ is lost –> hypo K
- Patients are also hypercalcaemic (+ hypocalciuric) –> chondrocalcinosis. Why?
- Theory 1: During contraction of ECV, PT cells are alerted to increase Na+ reabsorption. Passively, Ca2+ is driven down the electrochemical gradient, hence higher Ca2+ levels in the body
- Theory 2: enhanced basolateral Na/Ca exchange due to decreased intracellular Na+ concentration, leading to increased apical Ca2+ entry through Trpty5
- Hypomagnesaemia also happens. Why?
- K+ deficiency –> increased passive Mg+ secretion
- ?reduced abundance of Mg channels in the apical membrane
To more minor part of the Gitelman syndrome
- CLCNKB gene mutation
- Also present in the TAL – overlap with Bartter Type 3
| Gitelman | Type 3 Bartter | |
| Age at diagnosis | Variable | Variable |
| Symptoms | Tetany
Chondrocalcinosis |
Variable |
| K+ | Low | Low |
| Mg2+ | Low | Normal |
| Urine Ca2+ | Low | High/Normal |
| Nephrocalcinosis | No | Sometimes |
| Gene | NCCT | CLCNKB |
Differences of Gitelman vs Type 3 Bartter
EAST Syndrome (Epilepsy, Ataxia, Sensorineural deafness, salt wasting renal Tubulopathy)
- Autosomal recessive
- KCNJ10 mutation – also found in brain (glial cells) and ear (inner ear).
- Similar presentation as Gitelman’s (renal wise)
And if you gain function of the NCC transporter:
Gordon Syndrome / Pseudohypoaldosteronism Type II
- salt sensitive hypertension
- hyperkalaemia
- normal anion gap metabolic acidosis – NAGMA
- normal renal function
- Mutation in 2 different genes :
- WNK 4 – loss of function
- responsible for inhibition of NCC –> loss of inhibition –> unregulated Na+ uptake.
- Leads to decreased Na+ delivery in the CD and reduced tubular electronegativity –> H+ and K+ not secreted
- Expansion of ECV –> Hypertension –> shut down aldosterone and RAS activity
- WNK 1 – negative regulator of WNK 4, hence mutation in WNK 1 is called gain of function
- WNK 4 – loss of function
And finally the Collecting Duct
There are 2 cell types:
- Principal Cells
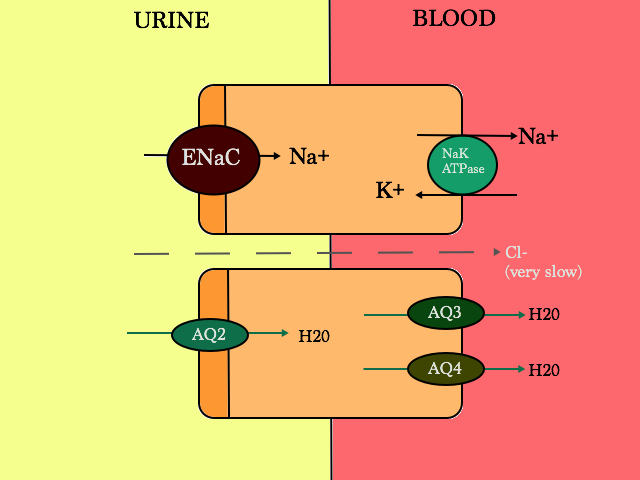
Properties of ENAC
- Na+ is reabsorped by ENAC and with that K+ is secreted out via ROMK (not shown here on the apical membrane)
- Regulated by aldosterone. Aldosterone is mediated by the mineralocorticoid receptor (MR) in the principal cells –> activated –> increase serine-threonine-kinase SGK1 (protein) –> increases more ENAC channels
- Inhibited by atrial natriuretic peptide (ANP). ANP is involved in cardiorenal homeostasis. High BP –> ANP activated via membrane-bound natriuretic peptide receptor-A –> generates second messenger cGMP –> down regulates ENAC/inhibits renin and aldosterone production
- can also be stimulated by vasopressin (ADH); but the net effect of vasopressin is to lower Na+ concentration
Properties of AQ receptors
- stimulated by vasopressin (ADH = antidiuretic hormone)
- water enters through AQ2 and exits via AQ3 and AQ4.
- water movement is driven via the tubulo-interstitium osmotic gradient
Problems with over activation of ENAC:
Liddle Syndrome
- Autosomal dominant
- Mutations in SCNN1B or SCNN1G – both genes code for breakdown of ENAC
- ENAC ++++ –> +++ Na reabsorption and +++ K secretion (via ROMK)
- Become hypertensive and hypokalaemic
- H+ is loss –> metabolic alkalosis
- Aldosterone is deactivated –> mimics apparent mineralocorticoid excess syndrome (AME)
- Testing :
- urine shows low levels of renin, aldosterone and angiotensin
- urine cortisol:cortisone =1, much higher ratio in AME as AME patients make less cortisone
- Treatment : Block the ENAC receptor –> amiloride/trimethoprim
Special mention:
Apparent mineralocorticoid excess syndrome
- Autosomal recessive
- mutations in HSD11B2 gene – codes for 11 beta hydroxysteroid dehydrogenase type 2 (kidney isozyme expressed in principal cells) –> role is to inactivate cortisol by converting it to cortisone (less active metabolite).
- Loss of inactivation –> ++++ cortisol –> cross react and activate MR as it is non selective
- Hence act like aldosterone activation –> +++ ENAC –> hypertension, hypokalaemia, metabolic a and hypernatraemia.
- Low renin/angio/aldo levels
- Liquorice intake blocks 11 beta hydroxysteroid dehydrogenase activity too –> hence mimics AME
- Treatment : Block ENAC or MR —> spironolactone/amiloride
- To differentiate Liddle’s and AME –> Liddle’s responds to just ENAC blockade but AME will respond to MR and ENAC blockade (spironolactone blocks MR)
- Treatment 2 : dexamethasone/steroids to inhibit ACTH
Problems with AQ receptors;
Nephrogenic Diabetes Insipidus
- Autosomal dominant or recessive, X linked or acquired
- Aut dom/recessive:
- mutation of AQP2 gene – defect in the AQ receptor
- X-linked
- mutation of AVPR2 gene – mutation of the vasopressin receptor (90%)
- Acquired causes:
- Lithium
- Antibiotics/antifungals/antivirals/antineoplastic meds/tolvaptan/foscarnet/amphotericin
- Prolonged electrolyte deficiencies e.g. hypokalaemia, hypercalcaemia
- Pregnancy
- Protein malnutrition
- Tubular disease
- Sickle
- OBstructive uropathy
- Sjogren’s
- Amyloid
- Aut dom/recessive:
- affects AQ2 – complete or partial resistance to ADH –> no AQ2 to reabsorb water
- Severe polyuria and polydipsia
- Volume deplete hypernatraemia —> body wants to retain water –> high circulating ADH levels
- Signs & symptoms :
- Variable neurological deficit
- Calcification on brain imaging
- Hypernatraemia causes brain to shrink leading to intracerebral bleeds
- Functional obstructions
- constant/chronic large vols micturition –> patients hold urine in –> megacystis / hydroureter / hydronephrosis
- Variable neurological deficit
- Investigations :
- Water deprivation test – patients who are affected cannot concentrate their urine
- DDAVP – patients who respond to DDAVP has a cerebral DI
- Treatment :
- Low salt, low protein diet
- Thiazides +/- amiloride
- thiazides increases secretion of Na+ into the urine, and decreases osmolarity of the serum, helping break the excessive thirst –> drinking +++ –> polyuria cycle
- Causes mild dehydration/vol depletion –> Enhances PT Na+/H20 reabsorption
- NSAIDS
- PGE2 antagonises ADH
- PG depletion enhances concentration
- Indomethacin > ibuprofen
- Desmopressin
- For non hereditary DI
2. Intercalated Cells (ICC) of which there are 2 types – α and β
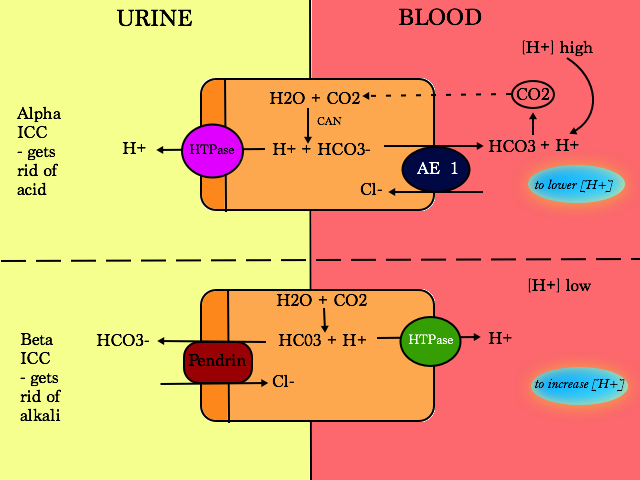
Involved in acid-base control
In the PT, HCO3 is reabsorbed.
In the ICC of the CD, HCO3 is generated to titrate acid and can also reabsorb HCO3 but to a lesser extent that the PT
Type A (α) ICC
- Acidifies urine (secrete H+)
- AE1 and HTPase is unregulated during metabolic acidosis
Type B (β) ICC
- Pendrin can transport Cl, Iodide and HCO3
- Pendrin is also present in the thyroid
- Pendrin over expression will lead to increased NaCl absorbtion
Type 1 Distal RTA – mutations in AE1
- unable to excrete the daily acid load, pH of urine >5.3
- Ammonia secretion is then impaired
- Proton accumulation –> normal anion gap metabolic acidosis + very low HCO3 levels
- will be buffered by phosphate from bone; bone adjustment with liberation of Ca2+ too –> osteomalacia
- Urine is very alkaline + buffering of acidosis + Ca2+ from bone –> hypercalciuria –> nephrocalcinosis and precipitation of urinary stones (which are Ca phosphate)
- Urine will have a negative charge –> to balance it, K+ is secreted –> hypokalaemia and hyperkalaeuria
Causes
Primary (isolated)
- AD – AE1 / Band-3 / SLC4A1 (CL-/HCO3- XC)
- AR – Apical H+-ATPase (sensorineural deafness)
Secondary
- Autoimmune disease
- Sjogrens
- Rheumatoids arthritis
- SLE
- Dysproteinaemias -myeloma
- Drugs
- Amphotericin
- Toluene toxicity
- Ifosfamide
- Lithium
- Hypercalciuric conditions (nephrocalcinosis)
- Hyperparathyroidism
- Idiopathic hypercalciuria
- Sarcoid
- Medullary sponge kidney
Clinical phenotype
- Hypercalciuria (acidosis ‘leaches’ bones)
- Hypocitraturia
- Nephrocalcinosis
- CaPO4 stones (alkaline urine)
- Propensity to UTI
- No Fanconi syndrome
- HCO3 variable (may be <10)
Diagnosis
- Urine pH always >5.5
- Caveats:
- Urea-splitting organism (exclude)
- AKI; low urine Na; prolonged ↓K
- Presence of nephrocalcinosis
- UAG POSITIVE (nb measure NH4+)
If doubt:
- Acidification test (NH4Cl)
- Frusemide and fludro (F’n’F test)
Special mention:
- Lithium is the same size and charge as Na+
- Li enters ENAC and disturbs the cell causing nephrogenic DI
- Amiloride can block ENAC and stop Lithium entering the cell
Type 4 Distal RTA
- occurs in the principal cell
- deficiency or resistance in the aldosterone receptor (MR) –> mimics aldosterone deficiency/hypoaldosteronism
- No ENAC –> no Na+ reabsorbtion –> urine is very ++ –> K+ is reabsorbed via ROMK —> hyperkalaemia
- Normal anion gap metabolic acidosis (hyperchloraemic met acidosis)
Causes
- Hereditary
- Primary pseudohypoaldosteronism type 1
- Renin and aldo elevated
- Sodium wasting
- Phenotype often mild in AD disease
- >Exacerbations (dehydration)
- AD (MR mutations)
- AR (ENAC mutations)
- Primary pseudohypoaldosteronism type 1
- Type 2 (Gordon syndrome) – AD
- Diabetes
- Adrenal failure
- HIV
- Drugs (block ENAC)
- NSAIDs
- ACE / ARB
- CNIs
- Heparin
- Amiloride
- Spiro
- Trimethoprim
- Structural renal disease
- Sickle
- Urinary tract obstruction
- Sickle
The Anion Gap
Positive : Na+ K+
Negative : HCO3- Cl –
Increased AG —
- not measuring H+ in:
- methanol
- ethylene glycol
- salicylates
- lactate
- ketones
or
2. HCO3 is low
- Barrter/Gitelman’s
- Diuretics
- Conn’s
Approaching Hypokalaemia
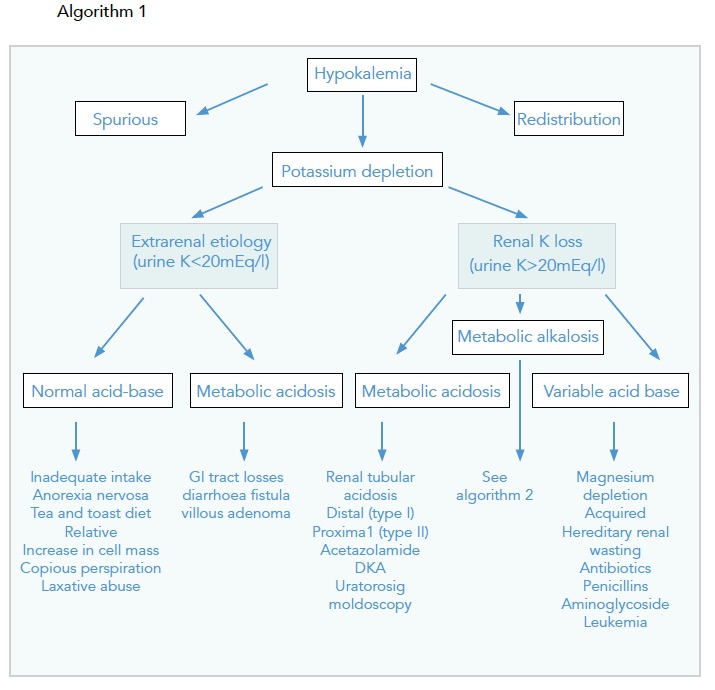
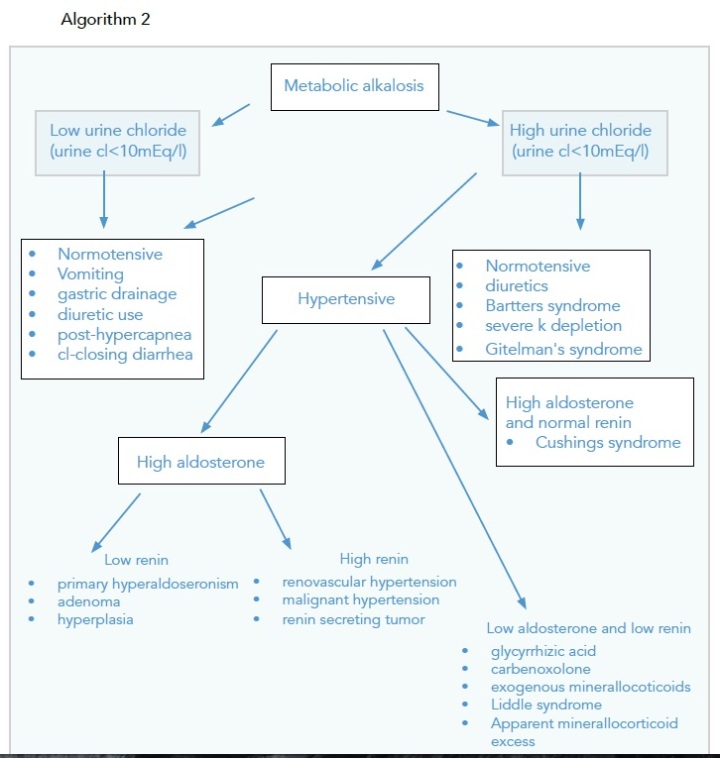
Special Mention
If NAGMA, measure the Urinary Anion Gap
- Na + K -Cl
- Measurement of NH4+
- In the presence of acidaemia, Kidney increases NH4 + excretion
- NH4+ is a counterion to Cl-
UAG NEGATIVE – large quantities of NH4+ in urine, distal acidification is intact
- All GI disturbances
- Proximal RTA
UAG POSITIVE – H+ secretion is impaired, or there is delivery of HC03 to the DT, or other ions eg ketones/hippurate is excreted
- Distal RTA
- T4 RTA
- Renal Failure
Not useful in polyuria / UNa < 20